


Conos: regroupement sur le réseau d'échantillons
-
Qu'est-ce que Conos?
CONOS est un package R pour câbler ensemble de grandes collections d'ensembles de données RNA-SEQ monocellulaire, ce qui permet à la fois l'identification de groupes de cellules récurrentes et la propagation des informations entre les ensembles de données dans des collections multi-échantillons ou à l'échelle de l'atlas. Il se concentre sur la cartographie uniforme des types de cellules homologues à travers les collections d'échantillons hétérogènes. Par exemple, les utilisateurs pourraient étudier une collection de dizaines d'échantillons de sang périphériques de patients cancéreux combinés avec des dizaines de témoins, ce qui comprend peut-être des échantillons d'un tissu apparenté tels que les ganglions lymphatiques. -
Comment ça marche?
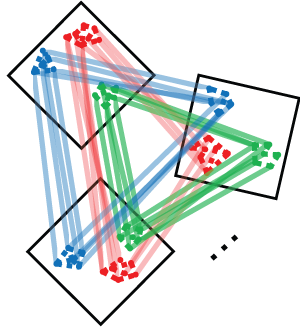
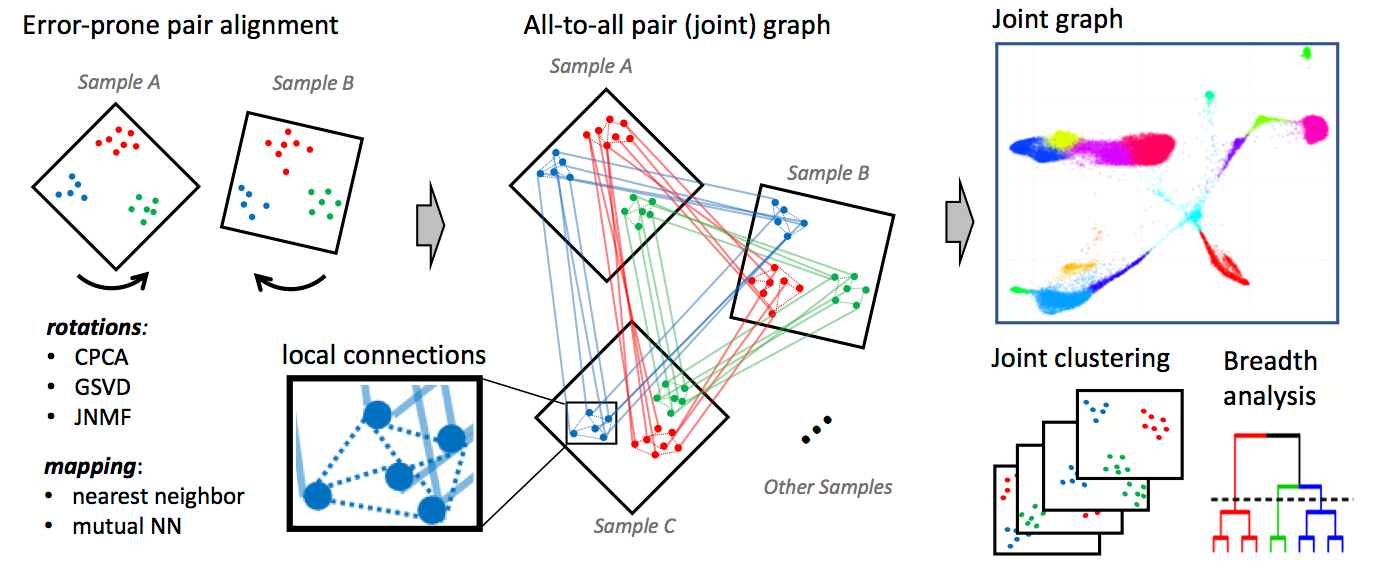
CONOS applique l'une des nombreuses méthodes sujettes aux erreurs pour aligner chaque paire d'échantillons dans une collection, établissant des liaisons de cellule à cellule inter-échantillon pondérées. Le graphique conjoint résultant peut ensuite être analysé pour identifier les sous-populations entre différents échantillons. Les cellules du même type auront tendance à se glisser les unes aux autres dans de nombreuses comparaisons par paires, formant des cliques qui peuvent être reconnues comme des grappes (communautés de graphiques).Le traitement des conos peut être divisé en trois phases:
- Phase 1: filtrage et normalisation Chaque ensemble de données individuel dans le panneau d'échantillonnage est filtré et normalisé à l'aide de packages standard pour le traitement unique: soit
pagoda2ouSeurat. Plus précisément, CONOS s'appuie sur ces méthodes pour effectuer un filtrage des cellules, la normalisation de la taille de la bibliothèque, l'identification des gènes surdispersés et, dans le cas de Pagoda2, normalisation de la variance. (Conos est robuste aux variations des procédures de normalisation, mais il est recommandé que tous les ensembles de données soient traités uniformément.) - Phase 2: Identifier plusieurs mappages inter-échantillons plausibles Conos effectue des comparaisons par paires des ensembles de données dans le panneau pour établir une cartographie initiale sujette aux erreurs entre les cellules de différents ensembles de données.
- Phase 3: Construction du graphique conjoint Ces bords inter-échantillons de la phase 2 sont ensuite combinés avec des bords intra-échantillon de poids inférieur pendant la construction du graphique conjoint. Le graphique conjoint est ensuite utilisé pour l'analyse en aval, y compris la détection de la communauté et la propagation des étiquettes. Pour une description complète de l'algorithme, veuillez vous référer à notre publication.
- Phase 1: filtrage et normalisation Chaque ensemble de données individuel dans le panneau d'échantillonnage est filtré et normalisé à l'aide de packages standard pour le traitement unique: soit
-
Que produit-il?
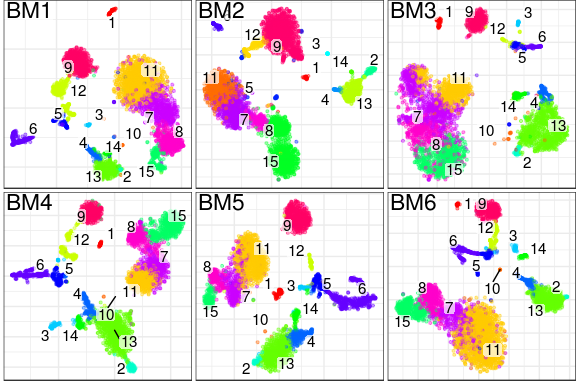
Essentiellement, Conos prendra un grand panneau potentiellement hétérogène d'échantillons et produira un regroupement de sous-populations cellulaires similaires d'une manière qui sera robuste à la variation inter-échantillon: -
Quels sont les avantages par rapport aux méthodes d'alignement existantes?
Conos est robuste à l'hétérogénéité des échantillons dans une collection, ainsi qu'au bruit. La capacité de résoudre la structure de sous-population plus fine s'améliore à mesure que la taille du panneau augmente.
Compte tenu d'une liste d'échantillons traités individuels (pl), le traitement des conos peut être aussi simple que ceci:
# Construct Conos object, where pl is a list of pagoda2 objects
con <- Conos$new(pl)
# Build joint graph
con$buildGraph()
# Find communities
con$findCommunities()
# Generate embedding
con$embedGraph()
# Plot joint graph
con$plotGraph()
# Plot panel with joint clustering results
con$plotPanel()
Pour voir plus de documentation sur la classe Conoscourir ?Conos.
Veuillez consulter les didacticiels suivants pour des exemples détaillés de la façon d'utiliser des conos:
Réglage de la résistance à l'alignement avec Conos:
Notez que pour l'intégration avec ScanPy, les utilisateurs doivent enregistrer des fichiers Conos sur le disque d'une session R, puis charger ces fichiers dans Python.
Enregistrer les conos pour ScanPy:
Chargez les fichiers Conos dans ScanPy:
Intégration de l'ARN-seq et de l'ATAC-Seq avec des Conos:
Exécution de la vitesse d'ARN sur un objet Conos
Tout d'abord, afin d'obtenir un tracé de vitesse d'ARN à partir d'un Conos Objet Vous devez utiliser le pipeline Drophest pour aligner et annoter vos mesures RNA-Seq à un cellule. Vous pouvez voir ce tutoriel et ce script shell pour voir comment cela peut être fait. Dans cet exemple, nous supposons spécifiquement que lors de l'exécution de Drop, vous avez utilisé le -V Option pour obtenir des estimations des comptes non épissés / épissés à partir du plus étourdi. Deuxièmement, vous avez besoin du package Velocyto.R pour l'estimation et la visualisation de la vitesse réelle.
Après avoir exécuté Drophest, vous devez avoir 2 fichiers pour chacun des échantillons:
sample.rds(matrice des comptes)sample.matrices.rds(3 matrices d'exons, d'introns et de lectures couvantes)
Le .matrices.rds Les fichiers sont les fichiers de vitesse. Chargez-les dans R dans une liste (même ordre que vous donnez à Conos). Charge, prétraitement et intégrer avec conos les matrices de comptage (.rds) comme vous le feriez normalement. Avant d'exécuter la vitesse, vous devez au moins créer une intégration et exécuter le clustering Leiden. Enfin, vous pouvez estimer la vitesse comme suit:
### Assuming con is your Conos object and cms.list is the list of your velocity files ###
library(velocyto.R)
# Preprocess the velocity files to match the Conos object
vi <- velocityInfoConos(cms.list = cms.list, con = con,
n.odgenes = 2e3, verbose = TRUE)
# Estimate RNA velocity
vel.info <- vi %$%
gene.relative.velocity.estimates(emat, nmat, cell.dist = cell.dist,
deltaT = 1, kCells = 25, fit.quantile = 0.05, n.cores = 4)
# Visualise the velocity on your Conos embedding
# Takes a very long time!
# Assign to a variable to speed up subsequent recalculations
cc.velo <- show.velocity.on.embedding.cor(vi$emb, vel.info, n = 200, scale = 'sqrt',
cell.colors = ac(vi$cell.colors, alpha = 0.5),
cex = 0.8, grid.n = 50, cell.border.alpha = 0,
arrow.scale = 3, arrow.lwd = 0.6, n.cores = 4,
xlab = "UMAP1", ylab = "UMAP2")
# Use cc=cc.velo$cc when running again (skips the most time consuming delta projections step)
show.velocity.on.embedding.cor(vi$emb, vel.info, cc = cc.velo$cc, n = 200, scale = 'sqrt',
cell.colors = ac(vi$cell.colors, alpha = 0.5),
cex = 0.8, arrow.scale = 15, show.grid.flow = TRUE,
min.grid.cell.mass = 0.5, grid.n = 40, arrow.lwd = 2,
do.par = F, cell.border.alpha = 0.1, n.cores = 4,
xlab = "UMAP1", ylab = "UMAP2")
Pour installer la version stable de Cran, utilisez:
install.packages('conos')
Pour installer la dernière version de conosutiliser:
install.packages('devtools')
devtools::install_github('kharchenkolab/conos')
Les dépendances sont héritées de Pagoda2. Notez que ce package a également l'IGRAPH de dépendance, qui nécessite que diverses bibliothèques s'installent correctement. Veuillez consulter les instructions d'installation de cette page pour plus de détails, ainsi que le GitHub Readme ici.
Pour installer des dépendances du système en utilisant apt-getUtilisez ce qui suit:
sudo apt-get update
sudo apt-get -y install libcurl4-openssl-dev libssl-dev libxml2-dev libgmp-dev libglpk-dev
Dépendances des distributions basées sur un chapeau rouge
Pour les distributions de chapeaux rouges en utilisant yumUtilisez la commande suivante:
sudo yum update
sudo yum install openssl-devel libcurl-devel libxml2-devel gmp-devel glpk-devel
À l'aide du Mac OS Package Manager Homebrew, essayez la commande suivante:
brew update
brew install openssl curl-openssl libxml2 glpk gmp
(Vous devrez peut-être courir brew uninstall curl afin de brew install curl-openssl Pour réussir.)
À partir de la version 1.3.1, conos devrait s'installer avec succès sur Mac OS. Cependant, s'il y a des problèmes, veuillez vous référer à la page Wiki suivante pour d'autres instructions sur l'installation conos avec Mac OS: Installation avec nous pour Mac OS
Si la configuration de votre système rend difficile l'installation conos nativement, un autre moyen d'obtenir conos L'exécution est via un conteneur Docker.
Note: Sur Mac OS X, la machine Docker a des limites de mémoire et de processeur. Pour le contrôler, veuillez vérifier les instructions pour CLI ou pour Docker Desktop.
Image docker prête à être
La distribution Docker a la dernière version et comprend également le package Pagoda2. Pour démarrer un conteneur Docker, installez d'abord Docker sur votre plate-forme, puis démarrez le pagoda2 conteneur avec la commande suivante dans le shell:
docker run -p 8787:8787 -e PASSWORD=pass pkharchenkolab/conos:latest
La première fois que vous exécutez cette commande, il téléchargera plusieurs grandes images, alors assurez-vous que vous avez une configuration d'accès Internet rapide. Vous pouvez ensuite pointer votre navigateur vers http: // localhost: 8787 / pour obtenir un environnement RStudio avec pagoda2 et conos installé (veuillez vous connecter à l'aide d'identification nom d'utilisateur =rstudiomot de passe =pass). Explorez l'option Docker – Mount pour permettre l'accès de l'image Docker à vos fichiers locaux.
Note: Si vous avez déjà téléchargé l'image Docker et que vous souhaitez la mettre à jour, veuillez extraire la dernière image avec:
docker pull pkharchenkolab/conos:latest
Bâtiment l'image docker du dockerfile
Si vous souhaitez créer une image par vous-même, téléchargez le dockerfile (disponible dans ce dépôt sous /docker) et exécuter vers la commande suivante pour la construire:
Cela créera une image Docker « Conos » sur votre système (soyez patient, car la construction pourrait prendre environ 30 à 50 minutes pour terminer). Vous pouvez ensuite l'exécuter en utilisant la commande suivante:
docker run -d -p 8787:8787 -e PASSWORD=pass --name conos -it conos
Si vous trouvez ce logiciel utile pour vos recherches, veuillez citer le document correspondant:
Barkas N., Petukhov V., Nikolaeva D., Lozinsky Y., Demharter S., Khodosevich K., & Kharchenko P.V.
Joint analysis of heterogeneous single-cell RNA-seq dataset collections.
Nature Methods, (2019). doi:10.1038/s41592-019-0466-z
Le package R peut être cité comme:
Viktor Petukhov, Nikolas Barkas, Peter Kharchenko, and Evan
Biederstedt (2021). conos: Clustering on Network of Samples. R
package version 1.5.2.

